January 25, 2012 By Steven Greer, MD
I have worn contact lenses for more than twenty years without any problems. But recently, my eyes began to disagree with them. In the course of investigating the possible reasons why, I uncovered some shocking inadequacies of the FDA reporting system meant to detect safety problems in medical devices, and also discovered ongoing problems in the way that Johnson and Johnson (JNJ) deals with medical device safety problems.
In October of 2011, I received a new batch of JNJ-made Acuvue Advance contact lenses (Silicone hydrogel lenses meant to be worn for two weeks), purchased from Utah supplier “1-800-CONTACTS”, which is now affiliated with Walmart in an “alliance” of distribution. I have used that supplier for this specific make and model since 2006. Shortly after receiving a new box in the mail, my eyes became inflamed. This was the first time that I had ever had trouble tolerating contact lenses.
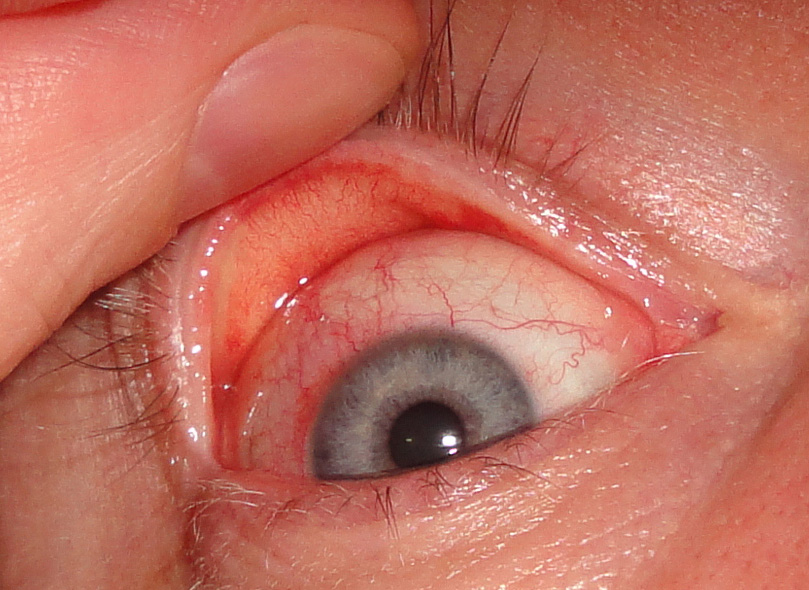
CLARE after JNJ Acuvue Advance contact lens wear
I do not sleep overnight in contact lenses, and when I would awake in the morning many hours after the lenses had been removed, excessive tear production overnight would form a crusty accumulation between my upper and lower lids. The inflammation would resolve approximately 24 hours after discontinuation of wearing the lenses. A catch-all term called CLARE (contact lens acute red eye) is applied to this condition.
In addition to the redness, I had developed a slight twitch in my upper right lid that could be induced consistently when I squinted and closed my lids forcefully. I have no neurological problems and have never before had such a problem. I am on no medications and received a clean bill of health from my last annual physical exam less than 12-months prior to the symptoms.
Being a medical doctor and also a healthcare analyst, I was well aware of the contact lens solution recalls back in 2006 due to manufacturing problems that caused serious eye infections and led to Buasch & Lomb collapsing and being sold to a private equity company. I promptly stopped wearing the contacts and went back to glasses as corrective lenses for myopia. I also proceeded to perform some medical experiments on myself to rule out certain variables.
I gave my eyes about two weeks of “time off” from the contact lenses, then tried wearing them again, using the same Alcon multipurpose solution. The CLARE inflammation returned. I switched to Bausch & Lomb “re-nu”, and the CLARE recurred just the same. I used the lenses straight from the blister-packages without adding any other lens solutions, and the CLARE recurred. All of this experimentation pretty much ruled out bacterial contamination of the contact lenses or lens solution. Although in rare cases, sometimes with counterfeit products, unopened blister packs have been contaminated with pseudomonas bacteria.
Medical doctor “MD” ophthalmology visits (As opposed to OD optometrists) are not covered under most health insurance plans unless there is an emergency room visit. Therefore, at this point of my saga, I unofficially consulted with two ophthalmologist friends of mine in Miami, and a third doctor with whom they put in contact with me up here in New York. Between all of them, their decades of training ranged from Harvard to Duke to Miami.
Based on my description of the problem, the three ophthalmology professors listed some possible etiologies for my CLARE. The contact lens fit (base curve) might have changed over time with long-term lens wear, thereby causing the lenses to be “too tight”, they all said. They also proposed that I might have suddenly developed an allergy even though the lens material had been tolerated by my eyes for many years. They all said that this was “common”.
Knowing medicine as I do, and how some popular diagnoses, not based on good clinical evidence, are nevertheless used when no better theory exists, I was skeptical of the theories being proposed by my friends. I kept searching for a more plausible etiology of my sudden inability to tolerate contact lenses.
Regarding my eyelid twitch, a neurology friend did not have an obvious explanation for it. He did say, however, that some of his residents had experienced the same problem while on long hospital shifts, and that their problems resolved after removing their contact lenses.
Based on my phone and email chats with my doctor friends, and the 1080i HD video of my eyes that I made for them, none of the doctors had included in their very unofficial “differential diagnoses” any manufacturing flaws of the contacts as the etiology of my CLARE. Moreover, the possibility that the lenses were counterfeit was something that never even crossed their minds. Most doctors completely trust that the FDA maintains safe and genuine supplies of drugs and devices.
I also contacted the head of the FDA’s CDRH medical device division, Dr. Jeffrey Shuren, in early December of 2011. I had interviewed him before on The Healthcare Channel. I did not request or expect his help with my personal matter, but was pleasantly surprised to see him escalate my problem to the medical officer in charge of contact lens regulations, Bernard Lepri.
The FDA initiated in December a review of their MAUDE database. This antiquated database system long predates Dr. Shuren’s service at the FDA, and has been criticized in numerous reports as woefully inadequate in detecting safety problems. Medical device malfunctions and medical complaints from patients rarely get reported to the FDA and seldom become recorded in the MAUDE database. Therefore, the statistical sensitivity of the MAUDE data are often very inadequate.
Approximately one month later, in January of 2012, the FDA replied to me with the results of their MAUDE database review. Since 2007, only 18 reports were found that remotely resembled my pattern of symptoms and were also related to my JNJ Acuvue Advance lenses. Those adverse events were mostly more severe and involved corneal ulcerations or blindness.
The FDA’s Bernard Lepri, an optometrist in the Division of Ophthalmic, Neurological and ENT Devices within CDRH, wrote to me the following, “Based upon the information you provided to FDA, we requested a MAUDE database MDR report on Acuvue contact lenses manufactured by Johnson & Johnson (JNJ). This report of consumer complaints totaled 180 for the period of time ranging from 2007 to December 20, 2011 for Acuvue contact lenses of various different brands and types. Of those 180 consumer complaints over five years, 18 reports were for Acuvue Oasys with Hydraclear. You are wearing Acuvue Advance with Hydraclear: Silicone Hydrogel Lenses; Daily Wear with 2-week replacement…. I reviewed these 18 individual reports. They all reported symptoms such as mucus discharge, sustained but temporary loss of vision associated with superficial punctate keratitis, iritis, corneal infiltrates, giant papillary conjunctivitis, papillae and follicles. Some of the reports also included diagnoses of epidemic keratoconjunctivitis which is not at all related to contact lens wear. These reports all indicated that the individuals wearing these lenses had contracted a variety of different forms of conjunctivitis, allergic, bacterial, and viral. These are not necessarily the result of a defect in the contact lenses they were wearing but most likely to exposure to pathogens and/or contact lens hygiene practices….The symptoms you described, while problematic and uncomfortable for you do not match the severity described in these 18 reports…. The description of your symptoms and the photograph of your eye (after wearing the lenses a couple of hours) is suspicious of CLARE (contact lens acute red eye) which has numerous etiologies: hypoxia, toxic effects from post-lens tear debris, mechanical irritation from a poor-fitting lens, dehydration of the tear lens while wearing lenses during sleep, solution hypersensitivity or toxicity or a reaction to bacterial toxins. This can occur even in seasoned contact lens wearers. While the occurrence may be acute, one could have been wearing the lenses for many years before it occurs. If this is indeed the problem you are experiencing, it may be necessary for you to change contact lens materials, brands, and/or care products individually or collectively….To date, there is no recall on the lenses in question.”
In short, the FDA reviewed the matter and concluded that there was no need to actively investigate further by reviewing the JNJ files that are more comprehensive than the “passive” MAUDE database. (I will elaborate on MAUDE later.) The FDA medical officer also seemed to give the JNJ and the Acuvue lens the benefit of the doubt as to the cause of the serious complications reported.
Back to November, 2011: My backups glasses were very old and I began the simple process, or so I thought, of buying new ones. I assumed they would cost just a few hundred bucks. After investigating online sources, and searching in states other than New York with lower costs of living, I was surprised to learn that the state-of-the-art plastic lenses, and quality frames, would set me back $1,000. Mind you, I have a simple non-progressive prescription. (Aside, radio personality Don Imus complained that his more complex RX set him back $3,000 for new glasses). With the help of my friends at the Bascom Palmer Eye Institute in Miami, I had the lenses made at cost, so I saved a few hundred dollars. For comfort reasons, I wanted saddle bridge rimless frames made by a German boutique designer, which I found at World Optical in Downtown New York (The service was excellent).
After approximately two months of wearing glasses and time off from contact lens use, I realized that my annoying right eyelid twitch had gone way. Whether or not the lens inflammation was the cause will never be known for sure.
In January of 2012, I had a new eye exam and contact lens fitting performed by an optometrist at World Optical. The base curve fit of my eyes had indeed not changed, ruling out the “too tight” contact lens theory as an etiology for my CLARE eye problems. My trial fitting of CooperVision Proclear contact lenses, and also of JNJ Acuvue Oasys lenses, went without a hitch and no CLARE was elicited. This sufficiently ruled out the theory of a new-onset allergy to silicone hydrogel materials.
I then did something very unusual. I went back and test wore the old Acuvue Advances that caused my problems in the first place. For the sake of science, and my own curiosity, I became a Guinea pig. Sure enough, my CLARE returned. At this point, it had been determined that I could wear other forms of JNJ and CooperVision silicone hydrogel lenses without trouble, and that my particular batch of Acuvue Advance received in October, 2011 could not be tolerated.
With all of the evidence now implicating my old Acuvue Advance lenses, I did a web search and saw that a different type of the JNJ Acuvue lens was recalled in Asia and Europe due to manufacturing process out of spec causing CLARE in users. More recently, CooperVision also issued a recall relating to out of spec coatings.
Something that I was then starting to consider as well was the possibility that my lenses were counterfeit. My experience as a healthcare analyst made me more suspicious of this than the average doctor, perhaps. I vaguely recalled in the business news reports of counterfeit contact lenses in Asia, and in the U.S. with 1-800-CONTACTS selling bogus CooperVision lenses.
If my offending lenses were not counterfeit, then possibly there was a manufacturing glitch. Not appreciated by most medical professionals in the optical field is that, despite being cheap and “disposable”, contact lenses are actually quite complicated to manufacture. Slight variations in specifications and tolerances can lead to unsafe products.
Also not well appreciated by medical doctors and optometrists is that contact lenses are approved for sale by the FDA without large randomized controlled trials in humans. Contact lenses are considered medium risk “Class 2” devices and are approved via the controversial 510(k) regulatory pathway obviating human clinical trials. Higher risk “Class 3” devices, such as coronary stents, require rigorous human clinical trials and the “PMA” regulatory pathway.
I spoke with one of the current FDA officers who evaluates contact lenses, a chemical engineer by training named Joseph Hutter, and he told me that the 510(k) for lenses is unique in that small cohort trials in humans are required when the materials are new or changed. Typically, the size of such a trial would be approximately 60 patients, and parameters such as slit lamp or CLARE are measured. Moreover, the current contact lens classification system used by the FDA, that was designed for lenses before the silicone hydrogel technology of today, was totally inadequate. After cases of blindness related to lens material and press coverage in 2006-07, the FDA developed a new system that was recently implemented (FDA slides).
In the case of the JNJ Acuvue Advance line that caused my problems, the product was found to be accumulating lipids and other “buildup” too rapidly and not allowing proper transmission of moisture to the cornea. In 2005, the Acuvue Oasys line made of a different silicone material, senofilcon A, was launched in order to address these problems. Also, according to a JNJ call center staffer, the Acuvue Advance line was modified in October of 2010 to address the excessive “buildup” and renamed Acuvue Advance “Plus”.
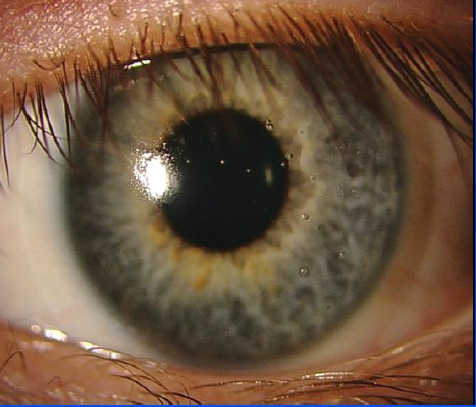
Lipid and calculi deposits on a contact lens. Photo from FDA slides.
The JNJ call center staff are given this explanation for callers as to why the “Plus” was added, “The surface was redesigned to eliminate lipid deposits and buildup.” When I pressed further as to what the “surface changes” were, I learned that the actual hydrophilic polymer embedded within the galyfilcon material was possibly “tweaked”.
Joe Hutter of the FDA contact lens group was nice enough to give me the actual patent number for this process, which is summarized on the USPTO site as, “A wettable silicone hydrogel made by including a high molecular weight hydrophilic polymer into the silicone hydrogel monomer mix is presented. The hydrophilic polymer is entrapped in the hydrogel with little or no covalent bonding between it and the hydrogel matrix.”
If JNJ had made changes to the Acuvue materials, they would have been obligated to file a report, at the least, with the FDA. I spoke at length with the FDA’s Joe Hutter and he searched all the databases at his disposal for any reports from JNJ that indicated JNJ made changes to the Acuvue Advance line. He found nothing since 2010 that would explain any chemical composition or manufacturing changes. There were only a few reports to the FDA from JNJ and they were on simple changes in methods for testing for raw materials.
The Acuvue Advance line has since been phased out and will no longer be sold as of this year. None of these extensive changes to the line that attempted to solve a patient safety matter were ever reported to the FDA as a formal recall, according to the best of the FDA investigator’s searches with whom we spoke. If any reports exist that we missed, they were not well flagged as “manufacturing changes to address patient complaints”. Was this another “stealth recall” for which JNJ has become infamous?
JNJ did not answer our direct questions as to whether they properly notified the FDA of material manufacturing changes, and whether this phasing out of the Acuvue Advance was a “stealth recall”. Instead, the head of corporate communications wrote this, “Dr. Greer, The Vistakon team has provided the following statement in response to your inquiry: “Vistakon regularly reviews our portfolio and makes business decisions about product offerings, with a goal of providing patients and physicians with new and advanced products. Both ACUVUE ADVANCE and ACUVUE ADVANCE Plus Brand Contact Lenses continue to be prescribed by eye care professionals to patients requiring vision correction.”
JNJ is currently under a consent decree of the department of Justice and FDA after it initiated “stealth recalls” of improperly manufactured over-the-counter Motrin, pediatric Tylenol, and other medications. In the medical device division, the company has been investigated for numerous cases of improperly recalling devices, most recently hip implants and diabetes pumps. Congress has strongly criticized the CEO of JNJ, Bill Weldon, during oversight hearings, and the former Deputy Commissioner of the FDA, Joshua Sharfstein, likely lost his job in relation to all of those aforementioned JNJ scandals as Rep. Darrell Issa became his nemesis.
At the time of this report, I have submitted my remaining supply of the offending Acuvue Advance contacts that caused my CLARE inflammation to be analyzed by the FDA and JNJ in order to determine whether they were made to spec and are even genuine JNJ products. The results of those analyses will be reported later, if we can obtain them.
Once all of the evidence gathered had implicated more and more the actual contact lenses as the culprit, as opposed to infectious contamination induced by me, allergies, or poor lens fit, I called 1-800-CONTACTS and was stunned by their handling of the complaint. They were happy to send me replacement contacts, but they did not seem eager to process my medical complaints and forward them on to the FDA or JNJ. Instead, they bounced me to the JNJ call center.
After speaking with numerous call center staff of 1-800-CONTACTS, I learned that the company has no internal database to track incoming complaints that allows a “central command” to spot patterns of problems, like the ones leading to the CooperVision recall. Complaints are kept in individual files. I wondered, what if 1-800-CONTACTS were receiving counterfeit lenses from China? How would they detect this if they did not even record complaints in a fashion that is accessible outside of individual customer charts? How would patterns of complaints about their products be flagged?
I then called the consumer hotline for JNJ contact lens matters and told them my story. They opened a case file and transferred me to a nurse, Gino Likins. I asked him whether he knew of any complaints similar to mine (i.e. CLARE) and he said, “We get them every day.” Knowing that the official FDA search of their MAUDE database since 2007 only revealed 18 complaints, and those were serious adverse events leading to corneal ulceration, etc, I asked him, “Do you report these to the FDA?”. The JNJ nurse said, “No…they are not serious enough”.
Given the fact that JNJ has issued recalls in Europe for Acuvue contact lenses recently, for coating problems that cause CLARE symptoms similar to my complaints, I was flabbergasted to hear the JNJ nurse openly explain that my symptoms were not serious enough to warrant reporting to the FDA. He went on to say, “Just because do not report complaints to the FDA does not mean we do not track them. We have an internal database and monitor trends all the time.”
It was then all becoming apparent to me why the FDA had only found 18 reports since 2007 in their MAUDE database related to Acuvue lenses, and that those were cases of severe eye injury. Neither JNJ, 1-800-CONTACTS, hospitals nor doctors are required to report to the FDA anything less than a “Death, serious injury, or malfunction”. Those three qualities are defined on page 17 and 18 of this FDA regulation. However, for any “complaint”, such as mine to the JNJ contact lens call center, the manufacturer is required to track those in a database and make them available to the FDA upon manufacturing plant inspections.
Usually, it is only after an outbreak of a serious safety problem related to a medical devices and subsequent FDA plant inspections that the full data are ever analyzed. This is what happened in 2006 with the cases of blindness from contact lens infections, and more recently with the JNJ Animus insulin pumps and JNJ metal-on-metal hip implant recalls. In all of those cases, The MAUDE database proved to be woefully inadequate as a source of early detection, particularly when the manufacturer fails to pass along the reports of medical device problems.
In 2008, to address the medical device safety monitoring inadequacies of the passive reporting MAUDE database, the FDA announced the Sentinel Initiative. It was designed as a more “active” database that allows the FDA to proactively search for problems rather than rely on random incoming reports, as MAUDE does. The FDA recently convened a meeting with the Brookings Institute to provide an update on the status of Sentinel, and Harvard’s Dr. Richard Platt was a lead presenter. FDA Commissioner Hamburg also gave an update report to congress in August of 2011.
Dr. Platt explained to me that Sentinel is still in the pilot program phase called Mini-Sentinel headed up by his Harvard Pilgrim Health Care Institute. Rather than a centralized database, it taps into a network of databases controlled by healthcare provider networks that form a “distributed database”. This addresses concerns that some doctors and administrators had in releasing personal information from medical records into a unsecured centralized “Big Brother” database that might violate HIPAA and discourage participation by doctors. Unlike MAUDE, manufacturers do not provide reports of drug or device complications to Sentinel. The data are derived from patient charts at hospital, health insurance databases (Figure 1), and other academic center sources. Also differing from MAUDE, Sentinel tracks drugs, vaccines, and medical devices, not just devices.
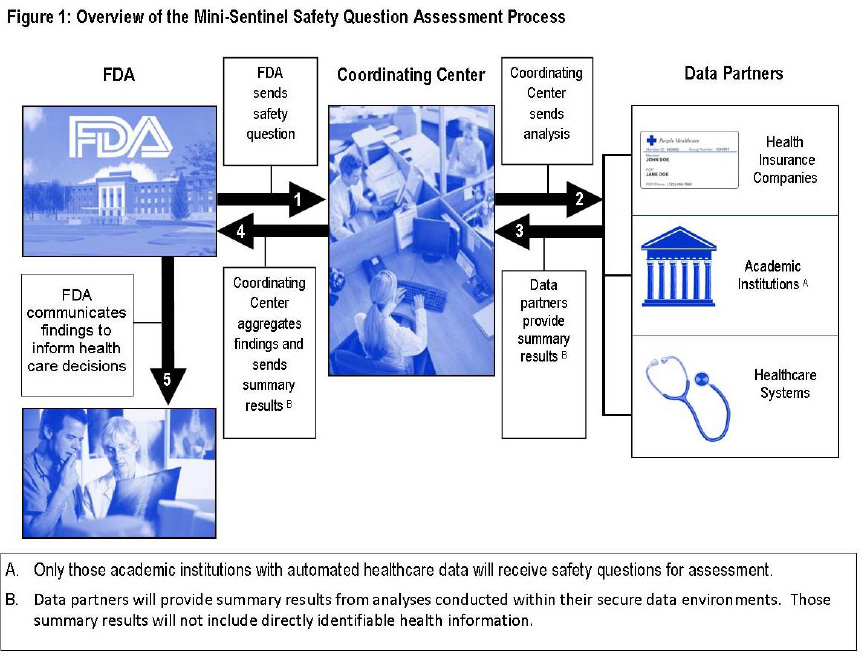 Based on the pilot Mini-Sentinel program, it does not seem any better than the current MAUDE database at detecting problems with contact lenses. The vast majority of health insurance plans do not cover optical care, and patients pay with their own cash to purchase contacts. Therefore, medical charts or insurance reports, two important sources of data for Sentinel, would not be collecting data on unsafe lenses.
Based on the pilot Mini-Sentinel program, it does not seem any better than the current MAUDE database at detecting problems with contact lenses. The vast majority of health insurance plans do not cover optical care, and patients pay with their own cash to purchase contacts. Therefore, medical charts or insurance reports, two important sources of data for Sentinel, would not be collecting data on unsafe lenses.
I asked Dr. Platt how products sold for cash, outside of insurance plans (e.g. contact lenses, cosmetic dermatology products like Juvederm or Botox, etc.) would be monitored under Sentinel. He explained, “Mini-Sentinel is currently only able to track medical products that are covered by the participating health plans, practices, or certain registries. It hasn’t tackled the items you mention.”
Whether the Sentinel database is ever allowed to be implemented, even with the inadequacies just mentioned, is highly uncertain. It is now four years after introduction and still in the “pilot phase” as the medical device industry thwarts any reforms that increase safety surveillance.
As a healthcare analyst, I am far more cynical of the FDA and medical device industry than the typical practicing doctor, and even I was amazed at how inadequate the safety monitoring systems are for the FDA. In fact, the class action tort lawyers do more to uncover patient complaints and safety problems, after the damage is done, than the FDA does in many cases.
Doctors should add to their textbook differential diagnoses the possibilities of adulterated or counterfeit drugs and devices, as well as malfunctioning devices. “Officially approved by the FDA” is not even remotely a guarantee that a device has been tested in humans and deemed safe.
Johnson and Johnson is the largest medical device maker in the world, and has an egregious track record of violating FDA regulations and the law. The billion in fines JNJ has paid out is just a rounding error for them and the cost of doing business. Criminal charges are rarely brought against a pharmaceutical executive, although a JNJ executive, fittingly, was one of the few to be convicted and jailed. But JNJ is by no means the lone offender. Stryker, Boston Scientific, Abbott, Medtronic, CooperVision, and scores of other medical device companies have all been investigated and prosecuted by the DOJ and FDA for failing to properly report safety problems and issue recalls.
Due to obvious incentives to conceal medical device malfunctions and prevent “recalls”, and all of the bad press that ensues, manufacturers cannot be trusted to reliably report safety trends to the FDA. Medical doctors remain as the last safety net in the medical device surveillance system, and are encouraged to report all patient complaints related to externally applied devices, such as contact lenses, as well as implantable devices. However, fears of malpractice lawsuits and paperwork hassle cause few doctors to report problems with medical devices.
This takes us full circle back to the federal agencies put in charge to protect the people against unsafe medical devices, which are the FDA taking the lead role and the Department of Justice as the muscle. But congressmen influenced by medical device lobbyists, such as the infamous and now-felon Tom DeLay, often stifle FDA safety regulations.
Senators Grassley, Blumenthal, and Kohl have recently introduced the “The Medical Device Patient Safety Act“. Based on a GAO report, the bill would “Improve post-market surveillance without stifling innovation or scrapping the current 510(k) approval system…and would help the FDA better identify when unsafe devices are on the market, and expedite the recall process once a serious problem is discovered.”
In the 2012 State of the Union speech, President Obama said, “That’s why we need smart regulations to prevent irresponsible behavior. Rules to prevent financial fraud or toxic dumping or faulty medical devices — these don’t destroy the free market. They make the free market work better.” After briefly courting the medical device industry in 2010, the Obama administration seems to be going back to emphasizing safety.